This paper gives a brief overview of protein chemistry, discusses the physical and chemical properties of protein glues using common models used in polymer science, describes the effect of fillers on these properties, and concludes with suggestions for possible future research topics in the area of gesso degradation and treatment.
The hide glues used as binders in gilding systems are formed from proteins that contain many polar and ionizable chemical groups. These groups participate in the linking of the glue molecules to the substrate and to themselves, thereby forming an interconnected network. This network has properties common to many cross-linked polymers; it is slightly elastic and can diffuse stress throughout its matrix. The addition of a bulking agent (whiting or gypsum) to the glue causes some changes in the chemical and physical properties of the protein network; most of these correlate to changes seen in filled cross-linked polymers.1 It is possible, then, to predict the behavior of a filled animal glue such as gesso by first considering the chemistry and physical properties of the components.
The chemical responsible for the adhesive properties of glue is collagen, the primary structural protein of animals. It exists as a long fibrous polymer molecule made up of amino acids in a complex, ordered configuration. A polymer is a molecule of high molecular weight, made up of many smaller molecules (monomers) that are linked end to end in a chainlike arrangement. The concept of a chain is a powerful model for many polymers; just as a real chain having 100 links is physically different from a pile of 100 separate links, so is a polymer chain composed of 100 monomers different from 100 separate molecules. If one link of the chain is pulled, the rest of the chain moves or stretches; the pile of separate links will be unaffected if a single link is moved. Also, if the chain is shaken, it will hold together, moving around within a restricted area, and may tend to get tangled. The pile of separate links will disperse on shaking, and the links will continue to move farther apart with no association between them. Thus, polymers are generally more flexible and more cohesive than the equivalent monomers.
Proteins are composed of amino acid monomeric units covalently linked together in specific sequence through peptide bonds:
The common amino acids that form the monomeric units of proteins are all carboxylic organic acids, which contain a primary amine (except two that have this nitrogen bound up in a ring) on the carbon next to the carboxyl carbon. A generalized structure of amino acid can be depicted as:
where "R" represents a variety of atoms that can be found attached to the basic structure, leading to the more than 20 individual amino acids found in nature (table 1). Their structures differ as complex variations of the amine-containing carboxylic acid theme. It is these variations that
Source: CL. Rose and D.W. von Endt, eds., Protein Chemistry for Conservators (Washington, D.C.: American institute for Conservation of Historic and Artistic Works, 1984), p. 5.
Fig. 1. Hydrogen bonding between protein chains, which can be broken by chemical or physical stress and reformed.
cause differentiation between protein types (bone protein versus silk protein, for example), and it is the combination of amines and acids that gives protein glues their particular proper-tie;. The simplified model of a protein chain in figure 1 shows the basic backbone of the chain with the interactive groups identified. The -NH- in the chains usually has a weak positive charge, and the=0 groups tend to have a weak negative charge. If these groups are close to each other, either on the same chain or on different chains, they will tend to associate, forming hydrogen bonds, but these bonds are easily broken and reformed. Hydrogen bonds, while weak compared to covalent bonds (the bonds that hold atoms together in a molecule), still produce strong cohesive forces between molecules. Hydrogen bonding makes it possible for glue molecules to interact strongly with appropriate substrates such as wood and to act as powerful adhesives.
Collagen, a basic "building block" in the construction of
animals, is referred to as a structural protein. It is fibrous in
nature and is a major component of the connective tissue that
sheaths muscles and attaches them to bone through tendons or that
attaches skeletal elements together through cartilage. It also forms
the bulk of the proteins found in hides and skin. When extracted
from hide, tendon, cartilage, and bones, collagen becomes the
primary component of glues. Fibrous proteins are well suited to
their task of support and connection. They are formed from very
long, thin fibers of amino acids covalently bonded in specific
sequence. This sequence gives collagen a specific shape and strength
that is a consequence of intramolecular hydrogen bonding. In the
1950s, Linus Pauling, Robert B. Corey, and H. R. Branson determined
that collagen exists as a molecule that is tightly coiled about
itself forming a secondary structure termed an  -helix.2 Nearly half the amino
acids of collagen are glycine and alanine, the smallest amino acids;
this causes the collagen molecule to coil in such a manner that the
small amino acids are in the center of the coil and the bulky, less
mobile ones.-for example, hydroxyproline- are on the outside (table 2). Hydroxyproline's rigid structure causes a
twist in the coil wherever it occurs. The -NH- and=0 groups of
glycine and alanine from one coil will then hydrogen bond with
similar sites on the other coils. The molecules bunch together in
groups of three, forming a larger coil that gives collagen fibers
their strength in living tissue. When collagen is heated in water,
it separates into the single coils, forming a solution (fig. 2a). As the solution cools, the collagen
chains try to reform the 3-coil structure but, like a misaligned
zipper, can line up only in sections. The unaligned parts of the
coils then align with parts of other coils. This pattern continues
until a network of the chains exists, held together by the
hydrogen-bonded, 3-coil links (fig. 2b).
-helix.2 Nearly half the amino
acids of collagen are glycine and alanine, the smallest amino acids;
this causes the collagen molecule to coil in such a manner that the
small amino acids are in the center of the coil and the bulky, less
mobile ones.-for example, hydroxyproline- are on the outside (table 2). Hydroxyproline's rigid structure causes a
twist in the coil wherever it occurs. The -NH- and=0 groups of
glycine and alanine from one coil will then hydrogen bond with
similar sites on the other coils. The molecules bunch together in
groups of three, forming a larger coil that gives collagen fibers
their strength in living tissue. When collagen is heated in water,
it separates into the single coils, forming a solution (fig. 2a). As the solution cools, the collagen
chains try to reform the 3-coil structure but, like a misaligned
zipper, can line up only in sections. The unaligned parts of the
coils then align with parts of other coils. This pattern continues
until a network of the chains exists, held together by the
hydrogen-bonded, 3-coil links (fig. 2b).
Table 2. A Comparison of Amino Acid Composition Expressed as Amino Acid Residues in a 100,000 Molecular Weight Protein
| Collagen | Casein | Albumin | Wool | |
| Glycine | 363 | 30 | 19 | 87 |
| Alanine | 107 | 43 | 35 | 46 |
| Valine | 29 | 54 | 28 | 40 |
| Leucine | 28 | 60 | 32 | 86 |
| Isoleucine | 15 | 49 | 25 | - |
| Serine | 32 | 60 | 36 | 95 |
| Threonine | 19 | 41 | 16 | 54 |
| Cystine | - | 2 | 1 | 49 |
| Methionine | 5 | 17 | 16 | 5 |
| Aspartic acid | 47 | 63 | 32 | 54 |
| Glutamic acid | 77 | 153 | 52 | 96 |
| Lysine | 31 | 61 | 20 | 19 |
| Hydroxylysine | 7 | - | - | - |
| Arginine | 49 | 25 | 15 | 60 |
| Histidine | 5 | 19 | 7 | 7 |
| Phenylalanine | 15 | 28 | 21 | 23 |
| Tyrosine | 5 | 45 | 9 | 26 |
| Tryptophan | - | 8 | 3 | 9 |
| Proline | 131 | 65 | 14 | 83 |
| Hydroxyproline | 107 | - | - | - |
SOURCE: C. L. Rose and D.W. von Endt, eds, Protein Chemistry for Conservators (Washington, D.C.: American Institute for Conservation of Historic and Artistic Works, 1984), pp. 8, 20.
Fig. 2. Conversion of tightly bonded, 3-coil strand of collagen, through heating and cooling, to a gelatin network.
The configuration is unique to collagen, since most other proteins-for example, albumin and casein-do not exist as fibers. These other proteins lack high amounts of the helix-producing amino acids; their significant amounts of glutamic acid and aspartic acids produce strong hydrogen bonds among molecules but do not produce any twists or coils in the chain. So while casein and albumen produce fair adhesives that will bond strongly to many substrates, these adhesives are brittle because they lack the built-in "springs" of the helix coils of hide glues.
Fig. 3. Limiting effect of filler molecules in network.
Thus, collagen-containing glues form a slightly elastic network of large molecules that are connected by hydrogen bonds. Much of the elasticity of the network comes from the coiled helix structure of the molecules, which is also reinforced by hydrogen bonds. The hydrogen bonds can be disturbed by humidifying the glue; water molecules will bond to sites that would normally be interchain linkages, and the network can be pulled apart more easily. This causes expansion in the bulk material. When the water is removed, the sites try to realign to hydrogen bond again, causing a shrinkage in the bulk material.
The commonest use of a filled animal glue is as a gesso,. a mixture of glue and whiting. Relating the chemistry of glue to the physical properties of gesso is simplified by using models. The most instructive model for a macromolecule with interconnections is a three-dimensional net. The source of the interconnections of the net differ with polymer type. With cured rubber, for example, the interconnections are the cross-links from vulcanization; in protein glue, the "cross-links" are a result of hydrogen bonding.3 The main difference is that rubber cross-links, while not easily broken, are also not easily reformed. An animal glue can, with some difficulty, be pulled apart enough to break the hydrogen bonds (or they may be disturbed by moisture or heat), but more bonds will form. These properties are well known, but the introduction of a filler will cause changes in the "self-healing" properties as well as the strength and elasticity of animal glues.
The introduction of a filler in the system reduces the mobility of the chains because the filler particles will keep the chains in a spread-out configuration. This will limit the "stretchiness" of the network (fig. 3). One expects the filled polymer to become stiffer as the amount of filler is increased because it takes more force to stretch the restricted network. Modulus (stiffness) is the slope of the stress-strain curve, which for the linear part of the curve is calculated as the stress (the amount of force required to produce some stretch) divided by the strain (the distance the object has stretched). So, if the two examples in figure 3 were stretched the same amount, and the filled sample required twice as much force to stretch it as the unfilled, the modulus would be twice that of the unfilled network. This increase in modulus is predicted by equations developed for filled polymers, such as the modified Kerner equation:
where Mc is the modulus of the composite (the filled
polymer), Mp is the modulus of the plain polymer,
Vf is the volume fraction of the filler, and A, B, and
 are
constants based on Mf, the modulus of the filler,
are
constants based on Mf, the modulus of the filler,  m, the maximum
packing function of the filler, and v, the volume change on
deformation of the plain polymer (fig.
4).4
m, the maximum
packing function of the filler, and v, the volume change on
deformation of the plain polymer (fig.
4).4
Fig. 4. Theoretical dependence of modulus of composite relative to unfilled polymer (Mc/Mp) on concentration of filler. Source: J. A. Manson and L. H. Sperling, Polymer Blends and Composites (New York: Plenum Press, 1976), p. 378.
Fig. 5. Theoretical curves for tensile strength of filled polymers. Source: Lawrence E. Nielsen, "Simple Theory of Stress-Strain Properties of Filled Polymers," Journal of Applied Polymer Science 10, no. 1 (January 1966): 99.
Although the modulus increases with the addition of filler, the ultimate tensile strength (the force required to pull the sample completely apart) decreases. As more filler is added, the protein network becomes more distended as it makes room for the filler particles. The filler particles distribute the strain unevenly along the protein chains; the protein network fails more easily when the stress is concentrated on a few chains at a lime. The equations used for filled polymers predict these results; in figure 5, the curve produced by assuming "no adhesion" between polymer and filler is the best model for a gesso system. Because fillers such as calcium carbonate and clay do not chemically interact with the polymers they fill, they are nonreinforcing fillers; they lack chemical adhesion necessary to increase the tensile strength of the polymer they fill. While a roughly surfaced filler can be adhered mechanically, the adhesion is weak compared to a chemical attraction between filler and glue.
When so much filler is added that no hydrogen bond links can be formed at all, the network has almost no tensile strength. This depends on the maximum distance between links. In a cross-linked polymer that has been filled beyond this maximum, there is a sharp drop in both the tensile modulus and the tensile strength; it is reasonable to expect that gesso would have a similar response. Since protein molecules are such long chains and have so many possible hydrogen bonding sites, however, we expect this maximum fraction to be much higher than those found in synthetic polymers (around 50% to 60%).5
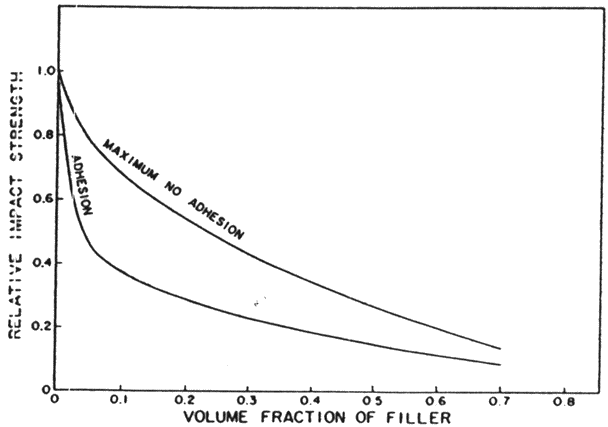
Fig. 6. Theoretical curves for the impact strength of filled polymers. Source: Lawrence E. Nielsen, "Simple Theory of Stress-Strain Properties of Filled Polymers," Journal of Applied Polymer Science 10, no. 1 (January 1966): 99.
If there were good adhesion between the glue and filler particles, we would probably see an increase in tensile strength with additional filler.6 This is a reasonable conclusion as the chemical links that would result from the protein chains bonding to the filler particles would replace the hydrogen bond links that the particles displaced. When such a system is subjected to stress, the links between the protein chains and the filler particles distribute the stress throughout the composite, rather than let it concentrate in a few chains. Good adhesion between filler and matrix, however, causes a decrease in the impact strength (fig. 6). Tests are necessary to determine if the loss in impact strength is worth the increase in tensile strength.
Fig. 7. Theoretical permeability to water of a glass-filled epoxy resin (Pr), relative to the unfilled polymer (Pr), according to the equation: PcPp = vp/(1+0.5vf). Source: J. A. Manson and E. H. Chiu, "Permeation of Liquid Water in a Filled Epoxy Resin," in Transport Phenomena through Polymer Films, ed. Charles A. Kumins (New York: Wiley Interscience, 1973), p. 105.
Although filling a glue with calcium carbonate should not affect the glue's reactivity with water, the filler will limit the expansion and contraction of the composite. In addition, we expect filled glue to be a better moisture barrier than unfilled glue. The addition of spherical filler particles to a polymer decreases its moisture permeability-the more filler, the less permeable (fig. 7). The models for this phenomenon are mostly based on the premise that filler dramatically increases the length of the path that the water molecule must traverse.7
The phenomenon poses a fundamental question in the investigation of the causes of gesso failure. Gesso exhibits "stress softening," a behavior seen in many filled plastics in which stress causes the chains to be moved irreversibly out of their network position around the filler particle. When the stress is released, the network partially returns to its original state; when stress is reapplied, less stress is required to stretch the filled plastic. As demonstrated by Marion Mecklenburg elsewhere in this volume, constrained gesso exhibits this behavior (often referred to as hysteresis) as a result of humidity changes. The upper layer of gesso generally has the most pigment and therefore is least permeable to moisture, which then raises the question of whether changes have an effect on the lower layers, or whether they are protected by the soft coat.
Study of filled animal glues raises new questions about possible causes of gesso failure, improvements in its properties, and considerations for its conservation, including the following:
1. Pigment variables-size, shape, and amount: What is the maximum amount of filler that can be added before the tensile modulus sharply decreases, and does it vary with chemical composition and particle size? Are there maximum and minimum useful particle sizes? What is the magnitude of the effect of the shape of the particles (for example, are smooth spheres better than the honeycomb shapes of diatoms)? Are changes in properties due to these variables large enough to cause problems with new fills (that is, must the gesso used for fills have similar pigment size and shape to the original)?
2. Adhesion variables: Can the adhesion between glue and filler be improved? If so, are the effects desirable? If the adhesion can be improved (by adding a bonding agent, perhaps), could the reaction be used as a consolidation procedure--that is, could the strength and cohesiveness of an old gesso be improved by adding an agent to increase the adhesion between the glue arid the filler?
3. Moisture permeation: How do the pigment and adhesion variables affect the moisture permeation? Does the soft coat protect the gesso layers from rapid humidity changes? Is gesso failure solely a function of the response of the underlying wood to humidity changes? How important is it to match the permeability of a fill to that of the original gesso?
The sheer number of questions remaining in these three areas alone indicates that, while some answers may be found in past research on filled polymers, more investigation is necessary to relate the theoretical to the practical.
The authors thank Conservation Science intern Nancie Ravenel for her help in preparing this manuscript.
1. For a more detailed introduction to protein chemistry and polymer science, see C. L. Rose and D. W. von Endt, eds., Protein Chemistry for Conservators (Washington, D.C.: American institute for Conservation of Historic and Artistic Works, 1984); James E. Mark and Burak Errnan, Rubberlike Elasticity: A Molecular Primer (New York: John Wiley and Sons, 1988).
2. Linus Pauling, Robert B. Corey, and H. R. Branson, "The Structure of Proteins: Two Hydrogen-Bonded Helical Configurations of the Polypeptide Chain," Proceedings of the National Academy of Sciences of the United States of America 37, no. 4 (April 1951): 205-11.
3. The interconnections in the collagen network are not covalent bonds and so are not true cross-links.
4. J. A. Manson and L. H. Sperling, Polymer Blends and Composites (New York: Plenum Press, 1976), p. 378.
5. M. W. Ranney, Reinforced Plastics and Elastomers, Recent Developments (Park Ridge, NJ.: Noyes Data Corp., 1977), pp. 300, 302.
6. L. Bateman, ed., The Chemistry and Physics of Rubber-Like Substances (New York: John Wiley and Sons, 1963), p. 311.
7. J. A. Manson and E. H. Chiu, "Permeation of Liquid Water in a Filled Epoxy Resin," in Transport Phenomena through Polymer Films, ed. Charles A. Kumins (New York: Wiley Interscience, 1973), pp. 95-108.
David W. von Endt received his Ph.D. in organic chemistry from Howard University in 1974. He is currently group leader of the organic chemistry division at the Conservation Analytical Laboratory, Smithsonian Institution. His present research centers on protein chemistry in the conservation and preservation of art and natural history specimens.
Mary T. Baker received her Ph.D. in polymer science from the University of Connecticut in 1986. She is currently a research chemist at the Conservation Analytical Laboratory, Smithsonian Institution. Her present research is on the conservation and preservation of modern polymeric materials.
Courtesy of Sound View Press, publisher of Guilded Wood: Conservation and History. This and other scholarly references on American art are available at http://www.falkart.com