Introduction to Colloid and Surface Chemistry. 4th. ed. Butterworth-Heinemann. Boston, 1992 ISBN 0 7506 1182 0. pp. 1-20.
Chapter 1.
Colloid science concerns systems in which one or more of the components has at least one dimension within the nanometre (10-9m) to micrometre (10-6m) range, i.e. it concerns, in the main, systems containing large molecules and/or small particles. The adjective 'microheterogeneous' provides an appropriate description of most colloidal systems. There is, however, no sharp distinction between colloidal and non-colloidal systems.
The range of colloidal systems of practical importance is vast, as is the range of processes where colloid/surface chemical phenomena are involved.
Examples of systems which are colloidal (at least in some respects) are:
| Aerosols | Foodstuffs |
| Agrochemicals | Ink |
| Cement | Paint |
| Cosmetics | Paper |
| Dyestuffs | Pharmaceuticals |
| Emulsions | Plastics |
| Fabrics | Rubber |
| Foams | Soil |
Examples of processes which rely heavily on the application of colloid/surface phenomena are:
| Adhesion | Ore flotation |
| Chromatography | Precipitation |
| Detergency | Road surfacing |
| Electrophoretic deposition | Sewage disposal |
| Emulsion polymerization | Soil conditioning |
| Food processing | Sugar refining |
| Grinding | Water clarification |
| Heterogeneous catalysis | Water evaporation control |
| Ion exchange | Water repellency |
| Lubrication | Wetting |
| Oil-well drilling |
As can be seen from the second of these lists, the existence of matter in the colloidal state may be a desirable or an undesirable state of affairs, and so it is important to know both how to make and how to destroy colloidal systems.
Colloid science is very much an interdisciplinary subject, albeit with certain areas of physics and physical chemistry most prominent. Owing to the complexity of most colloidal systems, the subject often cannot be treated readily with the exactness that tends to be associated with much of these major subject areas. It is probably a combination of this lack of precision and its interdisciplinary nature, rather than lack of importance, that has been responsible in the past for an unjustifiable tendency to neglect colloid science during undergraduate academic training.
Until the last few decades colloid science stood more or less on its own as an almost entirely descriptive subject which did not appear to fit within the general framework of physics and chemistry. The use of materials of doubtful compositions which put considerable strain on the questions of reproducibility and interpretation, was partly responsible for this state of affairs. Nowadays, the tendency is to work whenever possible with well-defined systems (e.g. monodispersed dispersions, pure surface-active agents, well-defined polymeric material) which act as models, both in their own right and for real life systems under consideration. Despite the large number of variables which are often involved, research of this nature coupled with advances in the understanding of the fundamental principles of physics and chemistry has made it possible to formulate coherent, if not always comprehensive, theories relating to many of the aspects of colloidal behaviour. Since it is important that colloid science be understood at both descriptive and theoretical levels, the study of this subject can range widely from relatively simple descriptive material to extremely complex theory.
The natural laws of physics and chemistry which describe the behaviour of matter in the massive and molecular states also, of course, apply to the colloidal state. The characteristic feature of colloid science lies in the relative importance which is attached to the various physicochemical properties of the systems being studied. As we shall see, the factors which contribute most to the overall nature of a colloidal system are:
Particle size
Particle shape and flexibility
Surface (including electrical) properties
Particle—particle interactions
Particle—solvent interactions
Colloidal systems may be grouped into three general classifications:
The particles in a colloidal dispersion are sufficiently large for definite surfaces of separation to exist between the particles and the medium in which they are dispersed. Simple colloidal dispersions are, therefore, two-phase systems. The phases are distinguished by the terms dispersed phase (for the phase forming the particles) and dispersion medium (for the medium in which the particles are distributed) — see Table 1.1. The physical nature of a dispersion depends, of course, on the respective roles of the constituent phases; for example, an oil-in-water (O/W) emulsion and a water-in-oil (W/O) emulsion could have almost the same overall composition, but their physical properties would be notably different (see Chapter 10).
Table 1.1 Types of colloidal dispersion
| Dispersed phase | Dispersion medium | Name | Examples |
| Liquid | Gas | Liquid aerosol | Fog, liquid sprays |
| Solid | Gas | Solid aerosol | Smoke, dust |
| Gas | Liquid | Foam | Foam on soap solutions, fire-extinguisher foam |
| Liquid | Liquid | Emulsion | Milk, mayonnaise |
| Solid | Liquid | Sol, colloidal suspension; paste (high solid concentration) | Au sol, AgI sol; toothpaste |
| Gas | Solid | Solid foam | Expanded polystyrene |
| Liquid | Solid | Solid emulsion | Opal, pearl |
| Solid | Solid | Solid suspension | Pigmented plastics |
Sols and emulsions are by far the most important types of colloidal dispersion. The term sol is used to distinguish colloidal suspensions from macroscopic suspensions; there is, of course, no sharp line of demarcation. When the dispersion medium is aqueous, the term hydrosol is usually used. If the dispersed phase is polymeric in nature, the dispersion is called a latex (pl. latices or latexes).
Foams are somewhat different in that it is the dispersion medium which has colloidal dimensions.
A characteristic feature of colloidal dispersions is the large area-to-volume ratio for the particles involved. At the interfaces between the dispersed phase and the dispersion medium characteristic surface properties, such as adsorption and electric double layer effects, are evident and play a very important part in determining the physical properties of the system as a whole. It is the material within a molecular layer or so of the interface which exerts by far the greatest influence on particle-particle and particle-dispersion medium interactions.
Despite this large area-to-volume ratio, the amount of material required to give a significant molecular coverage and modification of the interfaces in a typical colloidal dispersion can be quite small, and substantial modification of the overall bulk properties of a colloidal dispersion can often be effected by small quantities of suitable additives. For example, pronounced changes in the consistency of certain clay suspensions (such as those used in oil-well drilling) can be effected by the addition of small amounts of calcium ions (thickening) or phosphate ions (thinning)18.
Surface science is, therefore, closely linked with colloid science; indeed, colloid science is inevitably a part of surface science, although the reverse does not necessarily hold.
The surface or interfacial phenomena associated with colloidal systems such as emulsions and foams are often studied by means of experiments on artificially prepared flat surfaces rather than on the colloidal systems themselves. Such methods provide a most useful indirect approach to the various problems involved.
The terms lyophilic (liquid-loving) and lyophobic (liquid-hating) are frequently used to describe the tendency of a surface or functional group to become wetted or solvated. If the liquid medium is aqueous, the terms hydrophilic and hydrophobic are used.
Lyophilic surfaces can be made lyophobic, and vice versa. For example, clean glass surfaces, which are hydrophilic, can be made hydrophobic by a coating of wax; conversely, the droplets in a hydrocarbon oil-in-water emulsion, which are hydrophobic, can be made hydrophilic by the addition of protein to the emulsion, the protein molecules adsorbing on to the droplet surfaces.
This terminology is particularly useful when one considers the phenomenon of surface activity. The molecules of surface-active materials have a strong affinity for interfaces, because they contain both hydrophilic and lipophilic (oil-loving) regions.
The general usage of the terms ´lyophilic' and ´lyophobic' in describing colloidal systems is somewhat illogical. ´Lyophobic' traditionally describes liquid dispersions of solid or liquid particles produced by mechanical or chemical action; however, in these so-called ´lyophobic sols' (e.g. dispersions of powdered alumina or silica in water) there is often a high affinity between the particles and the dispersion medium - i.e. the particles are really lyophilic. Indeed, if the term ´lyophobic' is taken to imply no affinity between particles and dispersion medium (an unreal situation), then the particles would not be wetted and no dispersion could, in fact, be formed. 'Lyophilic' traditionally describes soluble macromolecular material; however, lyophobic regions are often present. For example, proteins are partly hydrophobic (hydrocarbon regions) and partly hydrophilic (peptide linkages, and amino and carboxyl groups).
The experimental procedures for determining particle size and shape can roughly be categorised, as follows:
Figure 1.1 Some model representations for non-spherical particles
Particle asymmetry is a factor of considerable importance in determining the overall properties (especially those of a mechanical nature) of colloidal systems. Roughly speaking, colloidal particles can be classified according to shape as corpuscular, laminar or linear (see, for example, the electron micrographs to Figure 3.2). The exact shape may be complex but, to a first approximation, the particles can often be treated theoretically in terms of models which have relatively simple shapes (Figure 1.1).
The easiest model to treat theoretically is the sphere, and many colloidal systems do, in fact, contain spherical or nearly spherical particles. Emulsions, latexes, liquid aerosols, etc., contain spherical particles. Certain protein molecules are approximately spherical. The crystallite particles in dispersions such as gold and silver iodide sols are sufficiently symmetrical to behave like spheres.
Corpuscular particles which deviate from spherical shape can often be treated theoretically as ellipsoids of revolution. Many proteins approximate this shape. An ellipsoid of revolution is characterised by its axial ratio, which is the ratio of the single half-axis a to the radius of revolution b. The axial ratio is greater than unity for a prolate (rugby-football-shaped) ellipsoid, and less than unity for an oblate (discus-shaped) ellipsoid.
Iron(III) oxide and clay suspensions are examples of systems containing plate-like particles.
High-polymeric material usually exists in the form of long threadlike straight or branched-chain molecules. As a result of inter-chain attraction or cross-finking (arising from covalent bonding, hydrogen bonding or van der Waals forces) and entanglement of the polymer chains, these materials often exhibit considerable mechanical strength and durability. This is not possible when the particles are corpuscular or laminar.
In nature, thread-like polymeric material fulfils an essential structural role. Plant life is built mainly from cellulose fibres. Animal life is built from linear protein material such as collagen in skin, sinew and bone, myosin in muscle and keratin in nails and hair. The coiled polypeptide chains of the so-called globular proteins which circulate in the body fluids are folded up to give corpuscular particles.
When particles aggregate together, many different shapes can be formed. These do not necessarily correspond to the shape of the primary particles.
Thread-like high-polymer molecules show considerable flexibility due to rotation about carbon-carbon and other bonds. In solution, the shape of these molecules alters continuously under the influence of thermal motion and a rigid rod model is therefore unsuitable. A better theoretical treatment is to consider the polymer molecules as random coils, but even this model is not completely accurate. Rotation about bonds does not permit complete flexibility, and steric and excluded volume effects also oppose the formation of a truly random configuration, so that, in these respects, dissolved linear polymer molecules will tend to be more extended than random coils. The relative magnitudes of polymer-polymer and polymer-solvent forces must also be taken into account. If the segments of the polymer chain tend to stick to one another, then a tighter than random coil, and possibly precipitation, will result; whereas a looser coil results when the polymer segments tend to avoid one another because of strong solvation and/or electrical repulsion.
Colloidal particles are usually solvated, often to the extent of about one molecular layer, and this tightly bound solvent must be treated as a part of the particle.
Sometimes much greater amounts of solvent can be immobilised by mechanical entrapment within particle aggregates. This occurs when voluminous flocculent hydroxide precipitates are formed. In solutions of long thread-like molecules the polymer chains may cross-link, chemically or physically, and/or become mechanically entangled to such an extent that a continuous three-dimensional network is formed. If all of the solvent becomes mechanically trapped and immobilised within this network, the system as a whole takes on a solid appearance and is called a gel.
Polydispersity and the averages
The terms relative molecular mass and particle size can only have well-defined meanings when the system under consideration is monodispersed - i.e. when the molecules or particles are all alike.
Figure 1.2 Particle diameter distribution for a polydispersed colloidal dispersion expressed (a) in histogram form, and (b) as a cumulative distribution
Colloidal systems are generally of a polydispersed nature - i.e. the molecules or particles in a particular sample vary in size. By virtue of their stepwise build-up, colloidal particle and polymer molecular sizes tend to have skew distributions, as illustrated in Figure 1.2, for which the Poisson distribution often offers a good approximation. Very often, detailed determination of relative molecular mass or particle size distribution is impracticable and less perfect experimental methods, which yield average values, must be accepted. The significance of the word average depends on the relative contributions of the various molecules or particles to the property of the system which is being measured.
Osmotic pressure, which is a colligative property, depends simply on the number of solute molecules present and so yields a number-average relative molecular mass:
Where ni is the number of molecules of relative molecular mass Mr,i.
In most cases the larger particles make a greater individual contribution to the property being measured. If the contribution of each particle is proportional to its mass (as in light scattering), a mass-average relative molecular mass or particle mass is given:
For any polydispersed system, Mr (mass average) > Mr (number average), and only when the system is monodispersed will these averages coincide. The ratio Mr (mass average)/Mr (number average) is a measure of the degree of polydispersity.
Basically, the formation of colloidal material involves either degradation of bulk matter or aggregation of small molecules or ions.
Dispersion of bulk material by simple grinding in a colloid mill or by ultrasonics does not, in general, lead to extensive subdivision, owing to the tendency of smaller particles to reunite (a) under the influence of the mechanical forces involved and (b) by virtue of the attractive forces between the particles. After prolonged grinding the distribution of particle sizes reaches an equilibrium. Somewhat finer dispersions can be obtained by incorporating an inert diluent to reduce the chances of the particles in question encountering one another during the grinding, or by wet-milling in the presence of surface-active material. As an example of the first of these techniques, a sulphur sol in the upper colloidal range can be prepared by grinding a mixture of sulphur and glucose, dispersing the resulting powder in water and then removing the dissolved glucose from the sol by dialysis.
A higher degree of dispersion is usually obtainable when a sol is prepared by an aggregation method. Aggregation methods involve the formation of a molecularly dispersed supersaturated solution from which the material in question precipitates in a suitably divided form. A variety of methods, such as the substitution of a poor solvent for a good one, cooling and various chemical reactions, can be utilised to achieve this end.
A coarse sulphur sol can be prepared by pouring a saturated solution of sulphur in alcohol or acetone into water just below boiling point. The alcohol or acetone vaporises, leaving the water-insoluble sulphur colloidally dispersed. This technique is convenient for dispersing wax-like material in an aqueous medium.
Examples of hydrosols which can be prepared by suitably controlled chemical reaction include the following:
The formation of a new phase during precipitation involves two distinct stages - nucleation (the formation of centres of crystallisation) and crystal growth - and (leaving aside the question of stability) it is the relative rates of these processes which determine the particle size of the precipitate so formed. A high degree of dispersion is obtained when the rate of nucleation is high and the rate of crystal growth is low.
The initial rate of nucleation depends on the degree of supersaturation which can be reached before phase separation occurs, so that colloidal sols are most easily prepared when the substance in question has a very low solubility. With material as soluble as, for example, calcium carbonate, there is a tendency for the smaller particles to dissolve (see page 68) and recrystallise on the larger particles as the precipitate is allowed to age.
The rate of particle growth depends mainly on the following factors:
Von Weimarn (1908) investigated the dependence on reagent concentration of the particle sizes of barium sulphate precipitates formed in alcohol-water mixtures by the reaction
Ba(CNS)2 + MgSO4 ->BaSO4 + Mg(CNS)2
At very low concentrations, c.10-4 to 10-3 mol dm-3, the supersaturation is sufficient for extensive nucleation to occur, but crystal growth is limited by the availability of material, with the result that a sol is formed. At moderate concentrations, c.10-2 to 10-1 mol dm-3, the extent of nucleation is not much greater, so that more material is available for crystal growth and a coarse filterable precipitate is formed. At very high concentrations, c.2 to 3 mol dm-3, the high viscosity of the medium slows down the rate of crystal growth sufficiently to allow time for much more extensive nucleation and the formation of very many small particles. Owing to their closeness, the barium sulphate particles will tend to link and the dispersion will take the form of a translucent, semi-solid gel.
Figure 1.3 The dependence of particle size on reagent concentration for the precipitation of a sparingly soluble material
The ageing of dispersions is discussed on page 68.
Aggregation methods usually lead to the formation of polydispersed sols, mainly because the formation of new nuclei and the growth of established nuclei occur simultaneously, and so the particles finally formed are grown from nuclei formed at different times. In experiments designed to test the validity of theories, however, there are obvious advantages attached to the use of monodispersed systems. The preparation of such systems requires conditions in which nucleation is restricted to a relatively short period at the start of the sol formation. This situation can sometimes be achieved either by seeding a supersaturated solution with very small particles or under conditions which lead to a short burst of homogeneous nucleation.
An example of the seeding technique is based on that of Zsigmondy (1906) for preparing approximately monodispersed gold sots. A hot dilute aqueous solution of HAuCl4 is neutralised with potassium carbonate and a part of the solute is reduced with a small amount of white phosphorus to give a highly dispersed gold sol with an average particle radius of c. 1 nm. The remainder of the HAuCl4 is then reduced relatively slowly with formaldehyde in the presence of these small gold particles. Further nucleation is thus effectively avoided and all of the gold produced in this second stage accumulates on the seed particles. Since the absolute differences in the seed particle sizes are not great, an approximately monodispersed sol is formed. By regulating the amount of HAuCl4 reduced in the second stage and the number of seed particles produced in the first stage, the gold particles can be grown to a desired size.
A similar seeding technique can be used to prepare monodispersed polymer latex dispersions by emulsion polymerisation (see page 17).
Among the monodispersed sols which have been prepared under conditions which lead to a short burst of homogeneous nucleation are (a) sulphur sols132, formed by mixing very dilute aqueous solutions of HCl and Na2S2O3; (b) silver bromide so1s133, by controlled cooling of hot saturated aqueous solutions of silver bromide; and (c) silver bromide and silver iodide so1s133, by diluting aqueous solutions of the complexes formed in the presence of excess silver or halide ions. In each case the concentration of the material of the dispersed phase slowly passes the saturation point and attains a degree of supersaturation at which nucleation becomes appreciable. Since the generation of dispersed phase material is slow, the appearance of nuclei and the accompanying relief of supersaturation is restricted to a relatively short period and few new nuclei are formed after this initial outburst. The nuclei then grow uniformly by a diffusion-controlled process and a sol of monodispersed particles is formed.
Figure 1.4 Formation of an approximately monodispersed sulphur sol by the slow reaction between Na2S2O3 and HCl in dilute aqueous solution
Various methods are also available for the preparation of monodispersed hydrous metal oxide sols19 and silica sols20.134.
Monodispersed polystyrene sols are used as calibration standards for electron microscopes, light scattering photometers, Coulter counters, particle sieves, etc. Monodispersed silica is used for antireflection lens coatings. Monodispersity (even at a modest level) can usefully be exploited in photographic film, magnetic devices, pharmaceutical preparations and catalysis.
Macromolecular chemistry covers a particularly wide field which includes natural polymeric material, such as proteins, cellulose, gums and natural rubber; industrial derivatives of natural polymers, such as sodium carboxymethyl cellulose, rayon and vulcanised rubber; and the purely synthetic polymers, such as polythene (polyethylene), Teflon (polytetrafluoroethylene), polystyrene, Perspex (poly (methyl methacrylate) ), terylene (poly (ethylene terephthalate) ) and the nylons, e.g. (poly (hexamethylene adipamide) ). Only brief mention of some of the more general aspects of polymerisation will be made. The reader is referred to the various specialised texts for details of preparation, properties and utilisation of these products.
High polymers contain giant molecules which are built up from a large number of similar (but not necessarily identical) units (or monomers) linked by primary valence bonds. Polymerisation reactions can be performed either in the bulk of the monomer material or in solution. A further technique, emulsion polymerisation, which permits far greater control over the reaction, is discussed on page 16.
There are two distinct types of polymerisation: addition polymerisation and condensation polymerisation.
Addition polymerisation does not involve a change of chemical composition. In general, it proceeds by a chain mechanism, a typical series of reactions being:
1. Formation of free radicals from a catalyst (initiator), such as a
peroxide. 2. Initiation: for example,
3. Propagation:
4. Termination. This can take place in several ways, such as reaction of the activated chain with an impurity, an additive or other activated chains, or by disproportionation between two activated chains.
A rise in temperature increases the rates of initiation and termination, so that the rate of polymerisation is increased but the average chain length of the polymer is reduced. The chain length is also reduced by increasing the catalyst concentration, since this causes chain initiation to take place at many mores points throughout the reaction mixture.
Condensation polymerisation involves chemical reactions between functional groups with the elimination of a small molecule, usually, water. For example,
If the monomers are bifunctional, as in the above example, then a linear polymer is formed. Terminating monofunctional groups will reduce the average degree of polymerisation. Polyfunctional monomers, such as glycerol and phthalic acid, are able to form branching points, which readily leads to irreversible network formation (see Chapter 9). Bakelite, a condensation product of phenol and formaldehyde, is an example of such a space-network polymer. Linear polymers are usually soluble in suitable solvents and are thermoplastic-i.e. they can be softened by heat without decomposition. In contrast, highly condensed network polymers are usually hard, are almost completely insoluble and thermoset-i.e. they cannot be softened by heat without decomposition.
A polymerisation method which is of particular interest to the colloid scientist is that of emulsion polymerisation.
In bulk polymerisation, processing difficulties are usually encountered unless the degree of polymerisation is sharply limited. These difficulties arise mainly from the exothermic nature of polymerisation reactions and the necessity for efficient cooling to avoid the undesirable effects associated with a high reaction temperature (see page 15). Even at moderate degrees of polymerisation the resulting high viscosity of the reaction mixture makes stirring and efficient heat transfer very difficult.
The difficulties associated with heat transfer can be overcome, and higher molecular weight polymers obtained, by the use of an emulsion system. The heat of polymerisation is readily dissipated into the aqueous phase and the viscosity of the system changes only slightly during the reaction.
A typical recipe for the polymerisation of a vinyl monomer would be to form an oil-in-water emulsion from:
| monomer (e.g. styrene) | 25-50 g |
| emulsifying agent (e.g. fatty acid soap) | 2-4 g |
| initiator (e.g. potassium persulphate) | 0.5-1 g |
| chain transfer agent (e.g. dodecyl mercaptan) | 0-0.2 g |
| water | 200 g |
Nitrogen is bubbled through the emulsion, which is maintained at c. 50-60°C for c. 4-6 h. The chain transfer agent limits the relative molecular mass of the polymer to c. 104, compared with c. 105-106 without it. The latex so formed is then purified by prolonged dialysis.
The mechanism of emulsion polymerisation is complex. The basic theory is that originally proposed by Harkins21. Monomer is distributed throughout the emulsion system (a) as stabilised emulsion droplets, (b) dissolved to a small extent in the aqueous phase and (c) solubilised in soap micelles (see page 89). The micellar environment appears to be the most favourable for the initiation of polymerisation. The emulsion droplets of monomer appear to act mainly as reservoirs to supply material to the polymerisation sites by diffusion through the aqueous phase. As the micelles grow, they adsorb free emulsifier from solution, and eventually from the surface of the emulsion droplets. The emulsifier thus serves to stabilise the polymer particles. This theory accounts for the observation that the rate of polymerisation and the number of polymer particles finally produced depend largely on the emulsifier concentration, and that the number of polymer particles may far exceed the number of monomer droplets initially present.
Monodispersed sols containing spherical polymer particles (e.g. polystyrene latexes22-24, 115) can be prepared by emulsion polymerisation, and are particularly useful as model systems for studying various aspects of colloidal behaviour. The seed sol is prepared with the emulsifier concentration well above the critical micelle concentration; then, with the emulsifier concentration below the critical micelle concentration, subsequent growth of the seed particles is achieved without the formation of further new particles.
Conventional filter papers retain only particles with diameters in excess of at least ìm and are, therefore, permeable to colloidal particles.
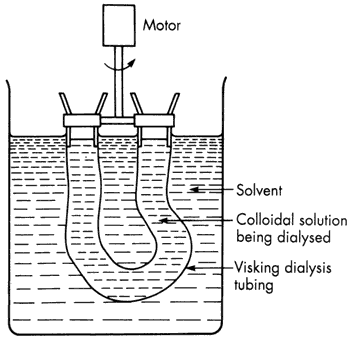
The use of membranes for separating particles of colloidal dimensions is termed dialysis. The most commonly used membranes are prepared from regenerated cellulose products such as collodion (a partially evaporated solution of cellulose nitrate in alcohol plus ether), Cellophane and Visking. Membranes with various, approximately known, pore sizes can be obtained commercially (usually in the form of ´sausage skins' or ´thimbles'). However, particle size and pore size cannot be properly correlated, since the permeability of a membrane is also affected by factors such as electrical repulsion when .the membrane and particles are of like charge, and particle adsorption on the filter which can lead to a blocking of the pores.
Figure 1.5 A simple dialysis set-up
Dialysis is particularly useful for removing small dissolved molecules from colloidal solutions or dispersions-e.g. extraneous electrolyte such as KNO3 from AgI sol. The process is hastened by stirring so as to maintain a high concentration gradient of diffusible molecules across the membrane and by renewing the outer liquid from time to time (Figure 1.5).
Ultrafiltration is the application of pressure or suction to force the solvent and small particles across a membrane while the larger particles are retained. The membrane is normally supported between fine wire screens or deposited in a highly porous support such as a sintered glass disc. An important application of ultrafiltration is the so-called reverse osmosis method of water desalination 25.
Another most valuable development of the ultrafiltration principle is the technique of gel permeation chromatography for the separation of the components of a polymeric sample and determination of the relative molecular mass distribution. The usual experimental arrangement involves the application of a pressure to force polymer solution through a chromatographic column filled with porous beads. The larger polymer molecules tend not to enter the pores of the beads and so pass through the column relatively quickly, whereas the smaller polymer molecules tend to diffuse through the pore structure of the beads and so take longer to pass through the column. The eluted polymer can be detected and estimated by measuring the refractive index of the emerging solution, and the relationship between retention time and relative molecular mass is determined by calibrating the apparatus with polymer fractions which have been characterised by other methods, such as osmotic pressure (see page 37), light scattering (see page 57) or viscosity (see page 251).
Figure 1.6 Electrodialysis
A further modification of dialysis is the technique of electrodialysis, as illustrated in Figure 1.6. The applied potential between the metal screens supporting the membranes speeds up the migration of small ions to the membrane surface prior to their diffusion to the outer liquid. The accompanying concentration of charged colloidal particles at one side and, if they sediment significantly, at the bottom of the middle compartment is termed electrodecantation.